Pegasus Workflows¶
Introduction¶
The Pegasus project encompasses a set of technologies that help workflow-based applications execute in a number of different environments including desktops, campus clusters, grids, and clouds. Pegasus bridges the scientific domain and the execution environment by automatically mapping high-level workflow descriptions onto distributed resources. It automatically locates the necessary input data and computational resources necessary for workflow execution. Pegasus enables scientists to construct workflows in abstract terms without worrying about the details of the underlying execution environment or the particulars of the low-level specifications required by the middleware. Some of the advantages of using Pegasus include:
-
Portability / Reuse - User created workflows can easily be run in different environments without alteration. Pegasus currently runs workflows on compute systems scheduled via HTCondor, including the OSPool, and other other systems or via other schedulers (e.g. XSEDE resources, Amazon EC2, Google Cloud, and many campus clusters). The same workflow can run on a single system or across a heterogeneous set of resources.
-
Performance - The Pegasus mapper can reorder, group, and prioritize tasks in order to increase the overall workflow performance.
-
Scalability - Pegasus can easily scale both the size of the workflow, and the resources that the workflow is distributed over. Pegasus runs workflows ranging from just a few computational tasks up to 1 million tasks. The number of resources involved in executing a workflow can scale as needed without any impediments to performance.
-
Provenance - By default, all jobs in Pegasus are launched via the kickstart process that captures runtime provenance of the job and helps in debugging. The provenance data is collected in a database, and the data can be summarized with tools such as pegasus-statistics or directly with SQL queries.
-
Data Management - Pegasus handles replica selection, data transfers and output registrations in data catalogs. These tasks are added to a workflow as auxiliary jobs by the Pegasus planner.
-
Reliability - Jobs and data transfers are automatically retried in case of failures. Debugging tools such as pegasus-analyzer help the user to debug the workflow in case of non-recoverable failures.
-
Error Recovery - When errors occur, Pegasus tries to recover when possible by retrying tasks, retrying the entire workflow, providing workflow-level checkpointing, re-mapping portions of the workflow, trying alternative data sources for staging data, and, when all else fails, providing a rescue workflow containing a description of only the work that remains to be done. Pegasus keeps track of what has been done (provenance) including the locations of data used and produced, and which software was used with which parameters.
As mentioned earlier in this book, OSG has no read/write enabled shared file system across the resources. Jobs are required to either bring inputs along with the job, or as part of the job stage the inputs from a remote location. The following examples highlight how Pegasus can be used to manage workloads in such an environment by providing an abstraction layer around things like data movements and job retries, enabling the users to run larger workloads, spending less time developing job management tools and babysitting their computations.
Pegasus workflows have 4 components:
- Site Catalog - Describes the execution environment in which the workflow will be executed.
- Transformation Catalog - Specifies locations of the executables used by the workflow.
- Replica Catalog - Specifies locations of the input data to the workflow.
- Workflow Description - An abstract workflow description containing compute steps and dependencies between the steps. We refer to this workflow as abstract because it does not contain data locations and available software.
When developing a Pegasus Workflow using the Python API, all four components may be defined in the same file.
For details, please refer to the Pegasus documentation.
Wordfreq Workflow¶
wordfreq is an example application and workflow that can be used to introduce
Pegasus tools and concepts.
The application is available on the OSG Access Points.
This example uses a custom container to run jobs. The container capability is provided by OSG (Containers - Apptainer/Singularity) and is used by setting HTCondor properties when defining your workflow.
Exercise 1: create a copy of the Pegasus tutorial and change the working directory to the wordfreq workflow by running the following commands:
$ git clone https://github.com/OSGConnect/tutorial-pegasus
$ cd tutorial-pegasus/wordfreq
In the wordfreq directory, you will find:
wordfreq/
├── bin
| ├── summarize
| └── wordfreq
├── inputs
| ├── Alices_Adventures_in_Wonderland_by_Lewis_Carroll.txt
| ├── Dracula_by_Bram_Stoker.txt
| ├── Pride_and_Prejudice_by_Jane_Austen.txt
| ├── The_Adventures_of_Huckleberry_Finn_by_Mark_Twain.txt
| ├── Ulysses_by_James_Joyce.txt
| └── Visual_Signaling_By_Signal_Corps_United_States_Army.txt
├── many-more-inputs
| └── ...
└── workflow.py
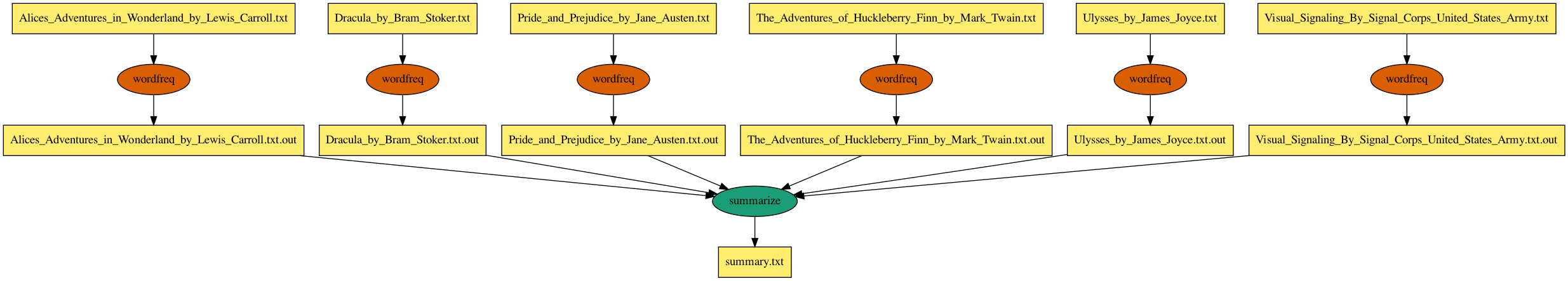
The inputs/ directory contains 6 public domain ebooks. The wordreq workflow uses the
two executables in the bin/ directory. bin/wordfreq takes a text file as input
and produces a summary output file containting the counts and names of the top five
most frequently used words from the input file. A wordfreq job is created for
each file in inputs/. bin/summarize concatenates the
the output of each wordfreq job into a single output file called summary.txt.
This workflow structure, which is a set of independent tasks joining into a single summary or analysis type of task, is a common use case on OSG and therefore this workflow can be thought of as a template for such problems. For example, instead of using wordfreq on ebooks, the application could be protein folding on a set of input structures.
When invoked, the workflow script (workflow.py) does the following major steps:
-
Generates a site catalog, which describes the execution environment in which the workflow will be run.
def generate_site_catalog(self): username = getpass.getuser() local = ( Site("local") .add_directories( Directory( Directory.SHARED_STORAGE, self.output_dir ).add_file_servers( FileServer(f"file://{self.output_dir}", Operation.ALL) ) ) .add_directories( Directory( Directory.SHARED_SCRATCH, self.scratch_dir ).add_file_servers( FileServer(f"file://{self.scratch_dir}", Operation.ALL) ) ) ) condorpool = ( Site("condorpool") .add_pegasus_profile(style="condor") .add_condor_profile( universe="vanilla", requirements="HAS_SINGULARITY == True", request_cpus=1, request_memory="1 GB", request_disk="1 GB", ) .add_profiles( Namespace.CONDOR, key="container_image", value="/cvmfs/singularity.opensciencegrid.org/htc/rocky:9" ) ) self.sc.add_sites(local, condorpool)In order for the workflow to use the container capability provided by OSG (Containers - Apptainer/Singularity), the following HTCondor profiles must be added to the condorpool execution site:
container_image=/cvmfs/singularity.opensciencegrid.org/htc/rocky:9. -
Generates the transformation catalog, which specifies the executables used in the workflow and contains the locations where they are physically located. In this example, we have two entries:
wordfreqandsummarize.def generate_transformation_catalog(self): wordfreq = Transformation( name="wordfreq", site="local", pfn=self.TOP_DIR / "bin/wordfreq", is_stageable=True ).add_pegasus_profile(clusters_size=1) summarize = Transformation( name="summarize", site="local", pfn=self.TOP_DIR / "bin/summarize", is_stageable=True ) self.tc.add_transformations(wordfreq, summarize) -
Generates the replica catalog, which specifies the physical locations of any input files used by the workflow. In this example, there is an entry for each file in the
inputs/directory.def generate_replica_catalog(self): input_files = [File(f.name) for f in (self.TOP_DIR / "inputs").iterdir() if f.name.endswith(".txt")] for f in input_files: self.rc.add_replica(site="local", lfn=f, pfn=self.TOP_DIR / "inputs" / f.lfn) -
Builds the wordfreq workflow. Note that in this step there is no mention of data movement and job details as these are added by Pegasus when the workflow is planned into an executable workflow. As part of the planning process, additional jobs which handle scratch directory creation, data staging, and data cleanup are added to the workflow.
def generate_workflow(self): # last job, child of all others summarize_job = ( Job("summarize") .add_outputs(File("summary.txt")) ) self.wf.add_jobs(summarize_job) input_files = [File(f.name) for f in (self.TOP_DIR / "inputs").iterdir() if f.name.endswith(".txt")] for f in input_files: out_file = File(f.lfn + ".out") wordfreq_job = ( Job("wordfreq") .add_args(f, out_file) .add_inputs(f) .add_outputs(out_file) ) self.wf.add_jobs(wordfreq_job) # establish the relationship between the jobs summarize_job.add_inputs(out_file)
Exercise 2: Submit the workflow by executing workflow.py.
$ ./workflow.py
Note that when Pegasus plans/submits a workflow, a workflow directory is created
and presented in the output. In the following example output, the workflow directory
is /home/ryantanaka/workflows/runs/ryantanaka/pegasus/wordfreq-workflow/run0014.
2020.12.18 14:33:07.059 CST: -----------------------------------------------------------------------
2020.12.18 14:33:07.064 CST: File for submitting this DAG to HTCondor : wordfreq-workflow-0.dag.condor.sub
2020.12.18 14:33:07.070 CST: Log of DAGMan debugging messages : wordfreq-workflow-0.dag.dagman.out
2020.12.18 14:33:07.075 CST: Log of HTCondor library output : wordfreq-workflow-0.dag.lib.out
2020.12.18 14:33:07.080 CST: Log of HTCondor library error messages : wordfreq-workflow-0.dag.lib.err
2020.12.18 14:33:07.086 CST: Log of the life of condor_dagman itself : wordfreq-workflow-0.dag.dagman.log
2020.12.18 14:33:07.091 CST:
2020.12.18 14:33:07.096 CST: -no_submit given, not submitting DAG to HTCondor. You can do this with:
2020.12.18 14:33:07.107 CST: -----------------------------------------------------------------------
2020.12.18 14:33:10.381 CST: Your database is compatible with Pegasus version: 5.1.0dev
2020.12.18 14:33:11.347 CST: Created Pegasus database in: sqlite:////home/ryantanaka/workflows/runs/ryantanaka/pegasus/wordfreq-workflow/run0014/wordfreq-workflow-0.replicas.db
2020.12.18 14:33:11.352 CST: Your database is compatible with Pegasus version: 5.1.0dev
2020.12.18 14:33:11.404 CST: Output replica catalog set to jdbc:sqlite:/home/ryantanaka/workflows/runs/ryantanaka/pegasus/wordfreq-workflow/run0014/wordfreq-workflow-0.replicas.db
[WARNING] Submitting to condor wordfreq-workflow-0.dag.condor.sub
2020.12.18 14:33:12.060 CST: Time taken to execute is 5.818 seconds
Your workflow has been started and is running in the base directory:
/home/ryantanaka/workflows/runs/ryantanaka/pegasus/wordfreq-workflow/run0014
*** To monitor the workflow you can run ***
pegasus-status -l /home/ryantanaka/workflows/runs/ryantanaka/pegasus/wordfreq-workflow/run0014
*** To remove your workflow run ***
pegasus-remove /home/ryantanaka/workflows/runs/ryantanaka/pegasus/wordfreq-workflow/run0014
This directory is the handle to the workflow instance and is used by Pegasus command line tools. Some useful tools to know about:
pegasus-status -v [wfdir]Provides status on a currently running workflow. (more)pegasus-analyzer [wfdir]Provides debugging clues why a workflow failed. Run this after a workflow has failed. (more)pegasus-statistics [wfdir]Provides statistics, such as walltimes, on a workflow after it has completed. (more)pegasus-remove [wfdir]Removes a workflow from the system. (more)
Exercise 3: Check the status of the workflow:
$ pegasus-status [wfdir]
You can keep checking the status periodically to see that the workflow is making progress.
Exercise 4: Examine a submit file and the *.dag.dagman.out files. Do these
look familiar to you from previous modules in the book? Pegasus is based on
HTCondor and DAGMan.
$ cd [wfdir]
$ cat 00/00/summarize_ID0000001.sub
...
$ cat *.dag.dagman.out
...
Exercise 5: Keep checking progress with pegasus-status. Once the workflow
is done, display statistics with pegasus-statistics:
$ pegasus-status [wfdir]
$ pegasus-statistics [wfdir]
...
Exercise 6: cd to the output directory and look at the outputs. Which is
the most common word used in the 6 books? Hint:
$ cd $HOME/workflows/outputs
$ head -n 5 *.out
Exercise 7: Want to try something larger? Copy the additional 994 ebooks from \ the many-more-inputs/ directory to the inputs/ directory:
$ cp many-more-inputs/* inputs/
As these tasks are really short, let's tell Pegasus to cluster multiple tasks
together into jobs. If you do not do this step, the jobs will still run, but not
very efficiently. This is because every job has a small scheduling overhead. For
short jobs, the overhead is obvious. If we make the jobs longer, the scheduling
overhead becomes negligible. To enable the clustering feature, edit the
workflow.py script. Find the section under Transformations:
wordfreq = Transformation(
name="wordfreq",
site="local",
pfn=self.TOP_DIR / "bin/wordfreq",
is_stageable=True
).add_pegasus_profile(clusters_size=1)
Change clusters_size=1 to clusters_size=50.
This informs Pegasus that it is ok to cluster up to 50 of the jobs which use the wordfreq executable. Save the file and re-run the script:
$ ./workflow.py
Use pegasus-status and pegasus-statistics to monitor your workflow. Using
pegasus-statistics, determine how many jobs ended up in your workflow and see
how this compares with our initial workflow run.
Variant Calling Workflow¶
This workflow is based on the Data Carpentry lesson Lesson Data Wrangling and Processing for Genomics.
This workflow downloads and aligns SRA data to the E. coli REL606 reference genome, and checks what differences exist in our reads versus the genome. The workflow also performs perform variant calling to see how the population changed over time.
The inputs are controlled by the recipe.json file. With 3 SRA inputs, the structure of the
workflow becomes:
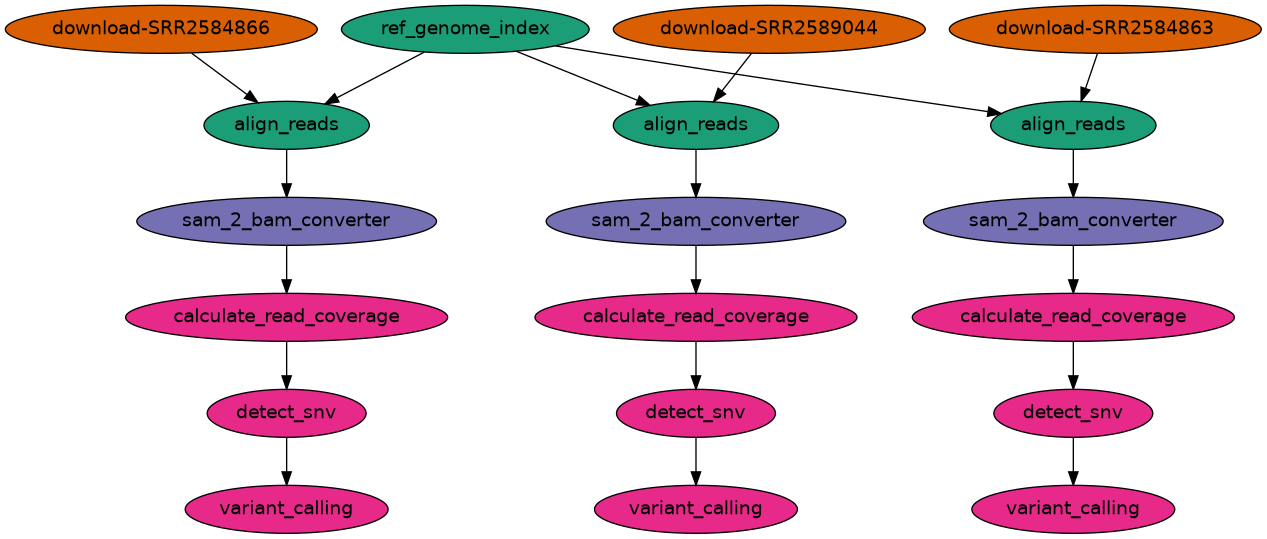
Rendering the workflow with data:
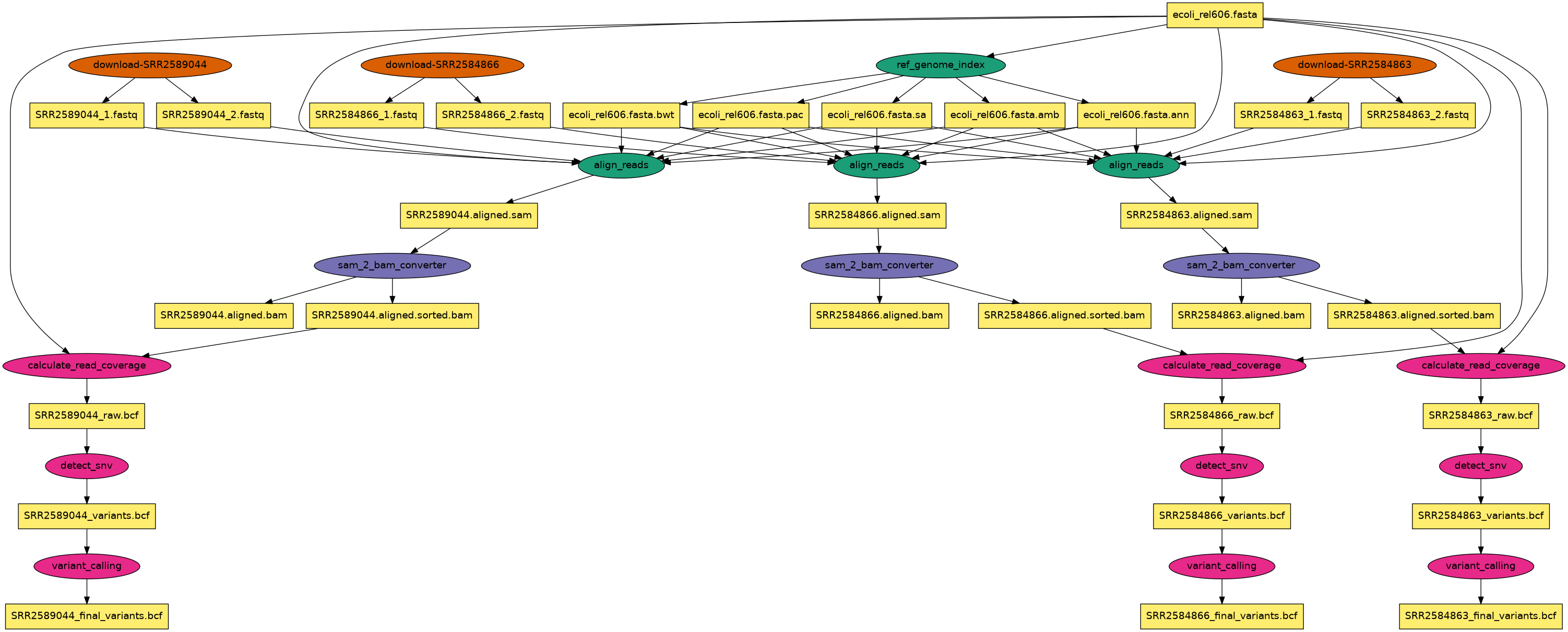
Compared to the wordfreq example, a difference is the use of
(OSDF)[https://osg-htc.org/services/osdf.html]
for intermediate data transfers/storage. Note the extra site in the site catalog:
osdf = (
Site("osdf")
.add_directories(
Directory(
Directory.SHARED_SCRATCH, f"{osdf_local_base}/staging"
).add_file_servers(
FileServer(f"osdf://{osdf_local_base}/staging", Operation.ALL)
)
)
)
Which is then referenced when planning the workflow:
self.wf.plan(
dir=str(self.runs_dir),
output_dir=str(self.output_dir),
sites=["condorpool"],
staging_sites={"condorpool": "osdf"},
OSDF is recommended for data sizes over 1 GB.
To plan the workflow:
$ ./workflow.py --recipe recipe.json
Getting Help¶
For assistance or questions, please email the OSG User Support team at [email protected] or visit the user documentation.